

Abstract
Concept. Targeted therapy of cancer typically focuses on inhibitors (e.g. tyrosine kinase inhibitors) that suppress oncogenic signaling below a minimum threshold required for survival and proliferation of cancer cells. Acute lymphoblastic leukemia (ALL) and B cell lymphomas originate from various stages of development of B cells, which unlike other cell types are under intense selective pressure. The vast majority of newly generated B cells are autoreactive and die by negative selection at autoimmunity checkpoints (AIC). Owing to ubiquitous encounter of self-antigen, autoreactive B cells are eliminated by overwhelming signaling strength of their autoreactive B cell antigen receptor (BCR). A series of recent findings suggests that, despite malignant transformation, AIC are fully functional in B cell malignancies. We propose that targeted engagement of AIC represents a previously unrecognized therapeutic opportunity to overcome conventional mechanisms of drug-resistance in pre-B ALL and other B cell malignancies.
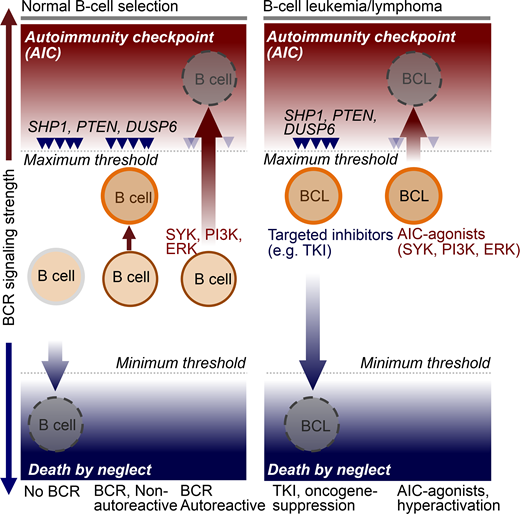
Results: Oncogenic drivers in B- cell malignancies function as mimics of B-cell receptor (BCR) signaling. Oncogenic activation of BCR-signaling represents the functional equivalent of positive selection during normal lymphocyte development. Addiction to survival and proliferation signals (or the equivalent of positive selection) is a common feature in many types of cancer. However, B-cell malignancies are unique in that they are also subject to an active negative selection process. B-cells expressing autoreactive BCRs or antibodies can cause systemic autoimmunity. As a safeguard against autoimmune diseases, lymphocyte development evolved autoimmunity checkpoints (AIC) to eliminate autoreactive clones. Owing to negative selection of autoreactive B-cells through AIC activation, lymphoid cells fundamentally differ in their signaling requirements from other cell types. Recent studies from our group showed that despite malignant transformation, B-cell leukemia and lymphoma cells are fully sensitive to negative selection and AIC-activation resulting (Chen et al., Nature 2015; Shojaee et al., Nature Med 2016; Chan et al., Nature 2017; Xiao et al., Cell 2018).
AIC-activation in various lymphoid malignancies is achievable by pharmacological hyperactivation of BCR-signaling above a maximum threshold (see Schematic below). Unlike other types of cancer, B-cell malignancies are uniquely susceptible to clonal deletion induced by hyperactive signaling from an autoreactive BCR. Hence, targeted AIC-activation can be leveraged for eradication of drug-resistant leukemia and lymphoma clones. Here, we propose a novel strategy to overcome drug-resistance in B-lymphoid malignancies based on targeted activation of autoimmunity checkpoints (AIC) for removal of autoreactive B-lymphocytes.
We have recently discovered that targeted hyperactivation of SYK, PI3K and ERK in B cell malignancies represents the functional equivalent of an autoimmunity checkpoint (AIC) for elimination of autoreactive clones among normal B cells. B cell tumors are uniquely vulnerable to AIC activation, suggesting that targeted activation of this checkpoint represents a novel strategy to induce cell death in otherwise drug-resistant B cell malignancies.
Conclusion: Normal B-cells are positively selected for BCR signaling of intermediate strength (moderate activation of SYK, PI3K and ERK). In the absence of a functional BCR, SYK, PI3K and ERK activity fall below a minimum threshold, resulting in death by neglect. Hyperactivation above maximum thresholds (e.g. autoreactive BCR) triggers negative selection and cell death via AIC-activation. Targeted therapy of cancer typically focuses on agents that suppress oncogenic signaling below a minimum threshold. Our results support a novel strategy to overcome drug-resistance in B-cell malignancies based on targeted activation of autoimmunity checkpoints (AIC) for removal of autoreactive cells.
No relevant conflicts of interest to declare.

